新闻中心

杏彩体育平台注册互联网软件设计app开发开发app流程图Nat Synth:神经
 2024-04-16 22:01:19
2024-04-16 22:01:19 浏览次数: 次
浏览次数: 次 返回列表
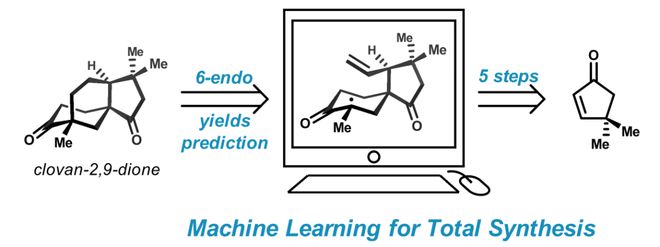
返回列表近日,耶鲁大学Newhouse课题组在Nature Synthesis上报道了以神经网络NNET模型预测关键反应产率,辅助实现三个Clovane天然产物的高效全合成,为机器学习模型在复杂小分子合成中的应用提供了范例。
复杂天然产物及药物分子(如紫杉醇和环孢素)的全合成工作被誉为有机化学领域的珠峰,杏彩体育注册其通常需要详细的逆合成分析和繁琐的实验验证,存在实验周期长、试错成本高等挑战。因为反应物化学结构上的小小改变可能对其化学反应性产生难以预料的影响;当关键反应的初步尝试失败时,研究人员通常需要修饰反应底物或调整反应顺序等不断迭代尝试;特别是当关键反应处于合成后期时,为合成带来更大风险,研究人员时常因关键反应失败而不得不放弃某一策略从头来过。
随着计算机技术和人工智能领域的飞速发展,如AlphaGo和ChatGPT的横空出世,人们开始担忧机器取代人类劳动力的时代正在加速到来,合成化学研究也因此迎来了新的机遇和挑战。近年来,计算机辅助的合成路线设计技术层出不穷(如Synthia 及Reaxys等商业化平台),然而它们在复杂体系中的实际应用仍需进一步改善(Nature 2020, 588, 83)。对于复杂小分子的高效合成,研究人员独特的创造性思维具有不可替代的地位。近日,耶鲁大学的Newhouse团队()提出了将人脑的创造性思维与计算机强大的虚拟分析能力相结合的“优势互补策略”,即以神经网络NNET模型预测关键反应(6-endo-tig自由基环化)的产率,指导合成路线设计和底物筛选,辅助实现了三个Clovane天然产物的高效全合成(5-8步),为机器学习模型指导复杂小分子的合成提供了范例。相关工作发表在Nature synthesis (DOI:10.1038/s41-0)
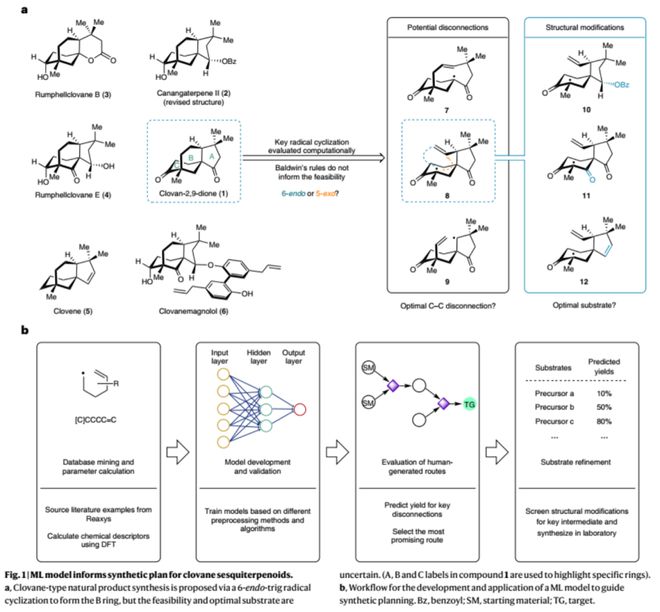
Clovane倍半萜广泛存在于各种海洋和陆地生物中,具有多样的生物活性。它们结构上大多具有独特的5/6/6三环桥环骨架及三个全碳季碳中心,为其合成带来一定挑战。该家族分子的化学合成已有大量研究,已知报道(10篇)均在合成前期引入或构建B环,如北京大学余志祥教授团队报道了特色的铑催化[3+2+1]环加成反应高效构建AB环系(Org. Lett. 2017, 19, 6040;Org. Lett. 2022, 24, 5902);四川大学刘波教授团队巧妙地以Carvone为原料经环丙烷化及酰化反应快速构建AB环,随后还原Aldol反应构建C环(Org. Lett. 2021, 23, 290)。区别于已知路线-endo-trig自由基环化为关键反应在合成后期构建B环的策略。自由基反应虽已被广泛应用于各类分子的合成中杏彩体育平台注册,但以类似转化构建6元桥环的例子并不多见,而以其竞争反应5-exo-trig为产物的报道居多,使得这一策略的可行性难以判断,Baldwin和 Beckwith等经验规则在此也无济于事。下载化学加APP到你手机,更加方便,更多收获。
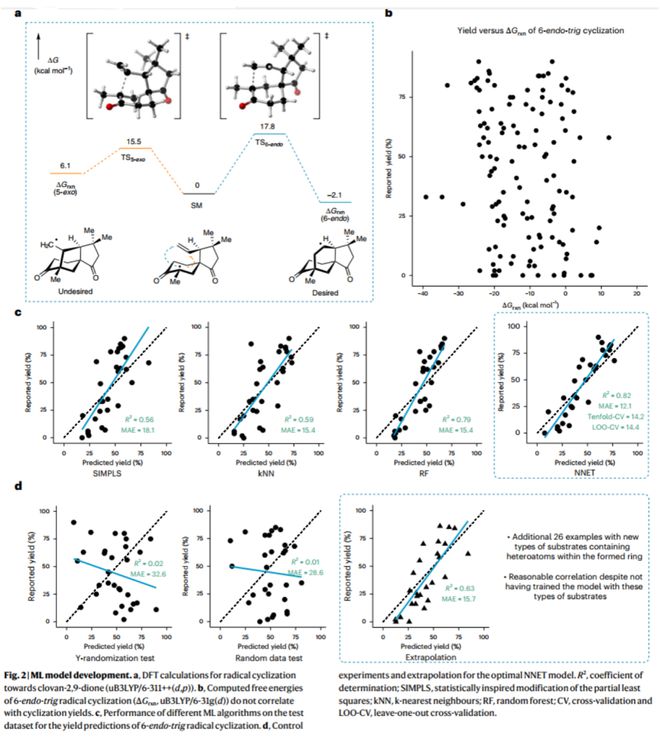
作者首先借助传统DFT计算方法分析该自由基反应过程,过渡态能垒ΔΔG‡的计算结果表明其竞争反应5-exo环化在动力学上更易发生;因6-endo环化作为热力学稳定产物,作者分析了120个已知6-endo-tig反应的能量变化(ΔGrxn)与产率之间的关系,但两者并无明显相关性(图2)。这些结果表明该化学反应产率受众多因素影响,因此作者设想以机器学习模型进行度、多参数的分析,以实现6-endo-trig环化反应产率的预测。
按照图1b所示的流程图,作者以Reaxys数据库中收录的自由基反应为研究样本,经分类和筛选、快速DFT计算(uB3LYP/6-31g(d))并提取相应物化参数(340个);随后经数据预处理(correlation及PCA)并用于模型的训练与验证。作者尝试训练了SIMPLS、kNN、RF等不同类型的模型,最终以神经网络模型(NNET)实现对6-end-trig自由基环化反应产率的预测(R2 = 0.82,MAE = 12%)。此外,作者进一步对模型进行了留一法交叉验证、Y随机化测试、随机数测试等验证,并以外推法对含杂原子的新型底物结构进行预测,结果表明模型可有效识别分子结构的化学信息。
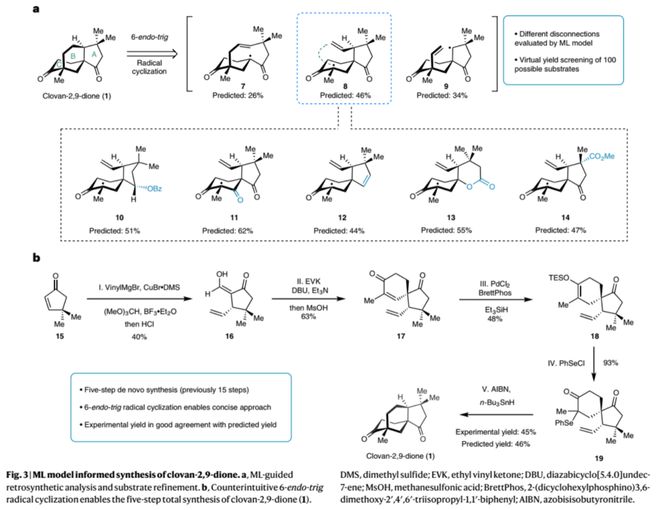
作者以训练好的神经网络NNET模型对不同逆合成切断方式(图3,7-9)以及100多个潜在底物结构进行了虚拟筛选,结果表明以前体8的6-endo-tig自由基环化反应具有较高可行性(预测产率46%),并为该家族分子的多样性合成挑选相应底物结构(10-14)。在模型指导下,作者从商业可得的原料15出发,经Michael加成、罗宾森环化、加氢硅烷化以及自由基环化等5步转化顺利实现了clovan-2,9-dione的高效全合成,极大提高了合成效率;其中6-endo-tig自由基环化的实验产率45%(6-endo:5-exo = 1:1)与模型预测的46%相一致。另一方面,通过对中间体17的CBS还原(Corey–Bakshi–Shibata reduction)及再氧化过程可实现该天然产物的不对称合成(8步)。
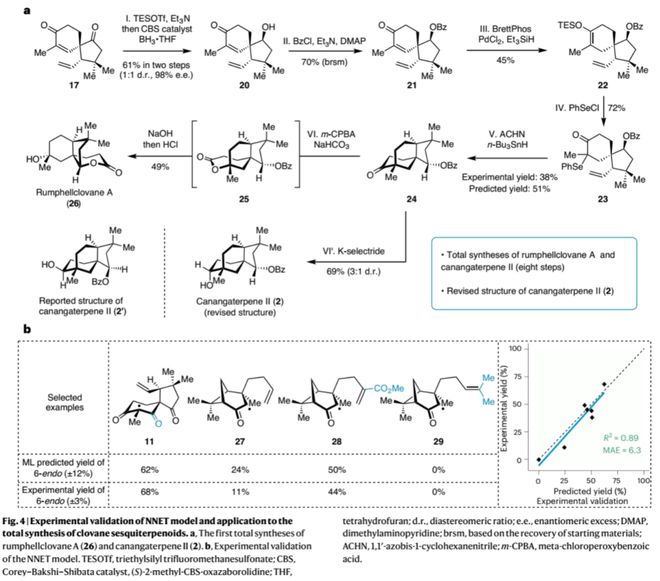
此外,在模型指导下,作者通过对中间体17进行结构修饰得到另一自由基反应前体10(硒化物23),顺利以38% 的收率实现6-endo自由基环化得到化合物24,进而经拜尔-维立格氧化重排或选择性还原反应分别实现了天然产物rumphellclovane A (26) 和canangaterpene II (2)的首次全合成(8步),并结合NMR计算修正了canangaterpene II(2)的立体结构。除此之外,作者还选取了不同类型的反应前体进行自由基反应实验,以验证不同取代基或骨架结构对6-endo-trig自由基反应产率的影响;所测试的7个反应的实验产率均与模型预测产率相一致,表现出极好的线%),从而证明了模型预测的准确性和可靠性;通过对比前体27与8的预测产率(24% Vs 46%)也可看出模型对化合物结构的敏感性。作者还指出,若以高精度的DFT计算分析100个潜在底物的反应路径及过渡态,在时间和算力上难以实现;而以训练好的机器学习模型可在一天内实现。
耶鲁大学Newhouse团队首次报道了“人机优势互补”策略在复杂小分子全合成中的应用,为其他类型预测模型的开发和应用提供了借鉴流程。该工作是在Timothy R. Newhouse教授指导下,由张鹏鹏博士与研究生Jungmin Eun等人合作完成,得到了耶鲁大学、Boehringer Ingelheim、Genentech以及美国NIH的大力支持。